Introduction to jsdmstan
jsdmstan.RmdJoint Species Distribution Models
Joint Species Distibution Models, or jSDMs, are models that model an entire community of species simultaneously. The idea behind these is that they allow information to be borrowed across species, such that the covariance between species can be used to inform the predictions of distributions of related or commonly co-occurring species.
In plain language (or as plain as I can manage) jSDMs involve the modelling of an entire species community as a function of some combination of intercepts, covariate data and species covariance. Therefore the change of a single species is related to not only change in the environment but also how it relates to other species. There are several decisions to be made in how to specify these models - the standard decisions on which covariates to include, whether each species should have its own intercept (generally yes) and how to represent change across sites - but also how to represent the covariance between species. There are two options for representing this species covariance in this package. First, the original way of running jSDMs was to model the entire covariance matrix between species in a multivariate generalised linear mixed model (MGLMM). However, more recently there have been methods developed that involve representing the covariance matrix with a set of linear latent variables - known as generalised linear latent variable models (GLLVM).
The jsdmstan package aims to provide an interface for fitting these models in Stan using the Stan Hamiltonian Monte Carlo sampling as a robust Bayesian methodology.
Underlying maths
Feel free to skip this bit if you don’t want to read equations, it is largely based on Warton et al. (2015). We model the community data for each site and taxon as a function of a species intercept, environmental covariates and species covariance matrix:
where is the link function, is the transpose of vector , and for each taxon , is an intercept and is a vector of regression coefficients related to measured predictors.
A site effect can also be added to adjust for total abundance or richness:
Multivariate Generalised Linear Mixed Models
The entire matrix of covariance between species is modelled in MGLMMs.
Fitting the entire covariance matrix means that the amount of time required to fit these models scales with the number of species cubed, and the data required scales with the number of species squared. This makes these models both computationally and data intensive.
Generalised Linear Latent Variable Models
In response to some of these issues in fitting MGLMMs, GLLVMs were developed in which is now specified as a linear function of a set of latent variables :
The latent variables are treated as random by assuming:
Treating the species covariance as pulling from a set of latent variables greatly reduces the computational time required to fit these models.
Relationship to environmental covariates
Within jsdmstan the response of species to environmental covariates
can either be unstructured (the default) or constrained by a covariance
matrix between the environmental covariates. This second option
(specified by setting beta_param = "cor") assumes that if
one species is strongly positively related to multiple covariates then
it is more likely that other species will either also be positively
related to all these covariates, or negatively related. Mathematically
this corresponds to:
Fitting a MGLMM
First we can use the in-built functions for simulating data according
to the MGLMM model - we’ll choose to simulate 15 species over 200 sites
with 2 environmental covariates. The species are assumed to follow a
Poisson distribution (with a log-link), and we use the defaults of
including a species-specific intercept but no site-specific intercept.
At the moment only default priors (standard normal distribution) are
supported. We can do this using either the jsdm_sim_data()
function with method = "mglmm" or with the
mglmm_sim_data() function which just calls
jsdm_sim_data() in the background.
nsites <- 75
nspecies <- 8
ncovar <- 2
mglmm_test_data <- mglmm_sim_data(N = nsites, S = nspecies,
K = ncovar, family = "pois")This returns a list, which includes the Y matrix, the X matrix, plus also the exact parameters used to create the data:
names(mglmm_test_data)
#> [1] "Y" "pars" "N" "S" "D" "K" "X"
dat <- as.data.frame(mglmm_test_data$X)Now, to fit the model we can use the stan_jsdm()
function, which interfaces to Stan through the rstan package. There are multiple
ways to supply data to the stan_jsdm() function, one is to
supply the data as a list with the appropriate named components (the
jsdm_sim_data() functions supply data in the correct format
already), the second way is to specify the Y and X matrices directly,
and the third way is to use a formula for the environmental covariates
and supply the environmental data to the data argument,
which is what we’ll use here:
mglmm_fit <- stan_jsdm(~ V1 + V2, data = dat, Y = mglmm_test_data$Y,
family = "pois", method = "mglmm", refresh = 0)If we print the model object we will get a brief overview of the type of jSDM and the data, plus if there are any parameters with Rhat > 1.01 or effective sample size ratio (Neff/N) < 0.05 then they will be printed:
mglmm_fit
#> Family: poisson
#> Model type: mglmm
#> Number of species: 8
#> Number of sites: 75
#> Number of predictors: 0
#>
#> Model run on 4 chains with 4000 iterations per chain (2000 warmup).
#>
#> Parameters with Rhat > 1.01, or Neff/N < 0.05:
#> mean sd 15% 85% Rhat Bulk.ESS Tail.ESS
#> cor_species[8,8] 1 0 1 1 1 7559 7522To get a summary of all the model parameters we can use
summary(), there are many parameters in these models so we
just include a few here:
summary(mglmm_fit, pars = "cor_species")
#> mean sd 15% 85% Rhat Bulk.ESS Tail.ESS
#> cor_species[2,1] 0.118 0.231 -0.109 0.354 1.003 1396 1985
#> cor_species[3,1] -0.225 0.155 -0.389 -0.064 1.003 2192 4145
#> cor_species[4,1] 0.459 0.098 0.355 0.563 1.002 2688 4582
#> cor_species[5,1] -0.185 0.190 -0.381 0.014 1.002 1917 3902
#> cor_species[6,1] 0.046 0.138 -0.096 0.188 1.001 2735 4392
#> cor_species[7,1] -0.111 0.303 -0.428 0.215 1.005 833 1530
#> cor_species[8,1] -0.046 0.296 -0.361 0.268 1.009 885 2190
#> cor_species[1,2] 0.118 0.231 -0.109 0.354 1.003 1396 1985
#> cor_species[2,2] 1.000 0.000 1.000 1.000 1.000 8125 NA
#> cor_species[3,2] 0.050 0.258 -0.216 0.318 1.000 2707 4354
#> cor_species[4,2] 0.384 0.242 0.147 0.623 1.002 1391 1335
#> cor_species[5,2] -0.133 0.280 -0.430 0.161 1.001 2576 4399
#> cor_species[6,2] -0.116 0.272 -0.397 0.167 1.000 2209 3810
#> cor_species[7,2] 0.000 0.311 -0.333 0.335 1.000 3232 5404
#> cor_species[8,2] -0.082 0.310 -0.415 0.253 1.000 3285 4954
#> cor_species[1,3] -0.225 0.155 -0.389 -0.064 1.003 2192 4145
#> cor_species[2,3] 0.050 0.258 -0.216 0.318 1.000 2707 4354
#> cor_species[3,3] 1.000 0.000 1.000 1.000 1.000 7678 NA
#> cor_species[4,3] -0.168 0.154 -0.330 -0.009 1.001 3564 5393
#> cor_species[5,3] 0.240 0.276 -0.050 0.532 1.000 2352 3713
#> cor_species[6,3] 0.148 0.227 -0.088 0.386 1.001 2672 4371
#> cor_species[7,3] 0.054 0.307 -0.274 0.383 1.001 2454 3902
#> cor_species[8,3] 0.040 0.312 -0.292 0.374 1.003 2707 4544
#> cor_species[1,4] 0.459 0.098 0.355 0.563 1.002 2688 4582
#> cor_species[2,4] 0.384 0.242 0.147 0.623 1.002 1391 1335
#> cor_species[3,4] -0.168 0.154 -0.330 -0.009 1.001 3564 5393
#> cor_species[4,4] 1.000 0.000 1.000 1.000 1.000 8123 NA
#> cor_species[5,4] -0.316 0.195 -0.522 -0.110 1.001 2347 4711
#> cor_species[6,4] -0.185 0.136 -0.325 -0.045 1.000 4293 5882
#> cor_species[7,4] 0.005 0.293 -0.302 0.316 1.002 1355 2678
#> cor_species[8,4] -0.130 0.295 -0.438 0.181 1.002 1315 2142
#> cor_species[1,5] -0.185 0.190 -0.381 0.014 1.002 1917 3902
#> cor_species[2,5] -0.133 0.280 -0.430 0.161 1.001 2576 4399
#> cor_species[3,5] 0.240 0.276 -0.050 0.532 1.000 2352 3713
#> cor_species[4,5] -0.316 0.195 -0.522 -0.110 1.001 2347 4711
#> cor_species[5,5] 1.000 0.000 1.000 1.000 1.000 8116 7643
#> cor_species[6,5] 0.372 0.198 0.158 0.583 1.001 3829 5532
#> cor_species[7,5] -0.017 0.314 -0.352 0.327 1.003 2314 4447
#> cor_species[8,5] 0.114 0.301 -0.214 0.434 1.000 3197 4494
#> cor_species[1,6] 0.046 0.138 -0.096 0.188 1.001 2735 4392
#> cor_species[2,6] -0.116 0.272 -0.397 0.167 1.000 2209 3810
#> cor_species[3,6] 0.148 0.227 -0.088 0.386 1.001 2672 4371
#> cor_species[4,6] -0.185 0.136 -0.325 -0.045 1.000 4293 5882
#> cor_species[5,6] 0.372 0.198 0.158 0.583 1.001 3829 5532
#> cor_species[6,6] 1.000 0.000 1.000 1.000 1.000 7519 NA
#> cor_species[7,6] 0.040 0.315 -0.301 0.377 1.001 1951 4334
#> cor_species[8,6] 0.162 0.289 -0.148 0.463 1.001 2117 3104
#> cor_species[1,7] -0.111 0.303 -0.428 0.215 1.005 833 1530
#> cor_species[2,7] 0.000 0.311 -0.333 0.335 1.000 3232 5404
#> cor_species[3,7] 0.054 0.307 -0.274 0.383 1.001 2454 3902
#> cor_species[4,7] 0.005 0.293 -0.302 0.316 1.002 1355 2678
#> cor_species[5,7] -0.017 0.314 -0.352 0.327 1.003 2314 4447
#> cor_species[6,7] 0.040 0.315 -0.301 0.377 1.001 1951 4334
#> cor_species[7,7] 1.000 0.000 1.000 1.000 1.001 8108 NA
#> cor_species[8,7] 0.006 0.320 -0.340 0.355 1.001 4288 6252
#> cor_species[1,8] -0.046 0.296 -0.361 0.268 1.009 885 2190
#> cor_species[2,8] -0.082 0.310 -0.415 0.253 1.000 3285 4954
#> cor_species[3,8] 0.040 0.312 -0.292 0.374 1.003 2707 4544
#> cor_species[4,8] -0.130 0.295 -0.438 0.181 1.002 1315 2142
#> cor_species[5,8] 0.114 0.301 -0.214 0.434 1.000 3197 4494
#> cor_species[6,8] 0.162 0.289 -0.148 0.463 1.001 2117 3104
#> cor_species[7,8] 0.006 0.320 -0.340 0.355 1.001 4288 6252
#> cor_species[8,8] 1.000 0.000 1.000 1.000 1.000 7559 7522To get a better overview of the R-hat and effective sample size we
can use the mcmc_plot() function to plot histograms of
R-hat and ESS.
mcmc_plot(mglmm_fit, plotfun = "rhat_hist")
#> `stat_bin()` using `bins = 30`. Pick better value `binwidth`.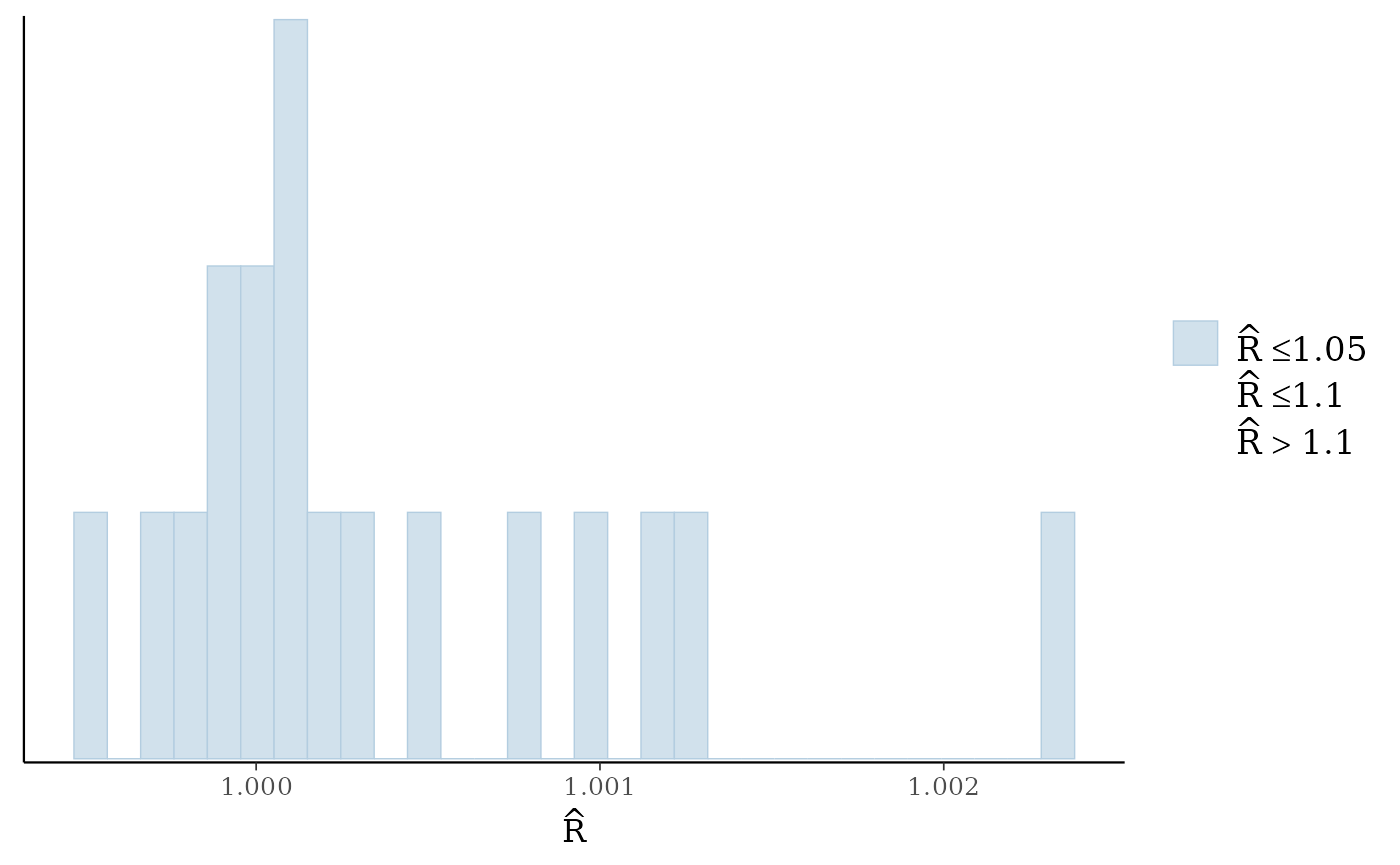
mcmc_plot(mglmm_fit, plotfun = "neff_hist")
#> Warning: Dropped 1 NAs from 'new_neff_ratio(ratio)'.
#> `stat_bin()` using `bins = 30`. Pick better value `binwidth`.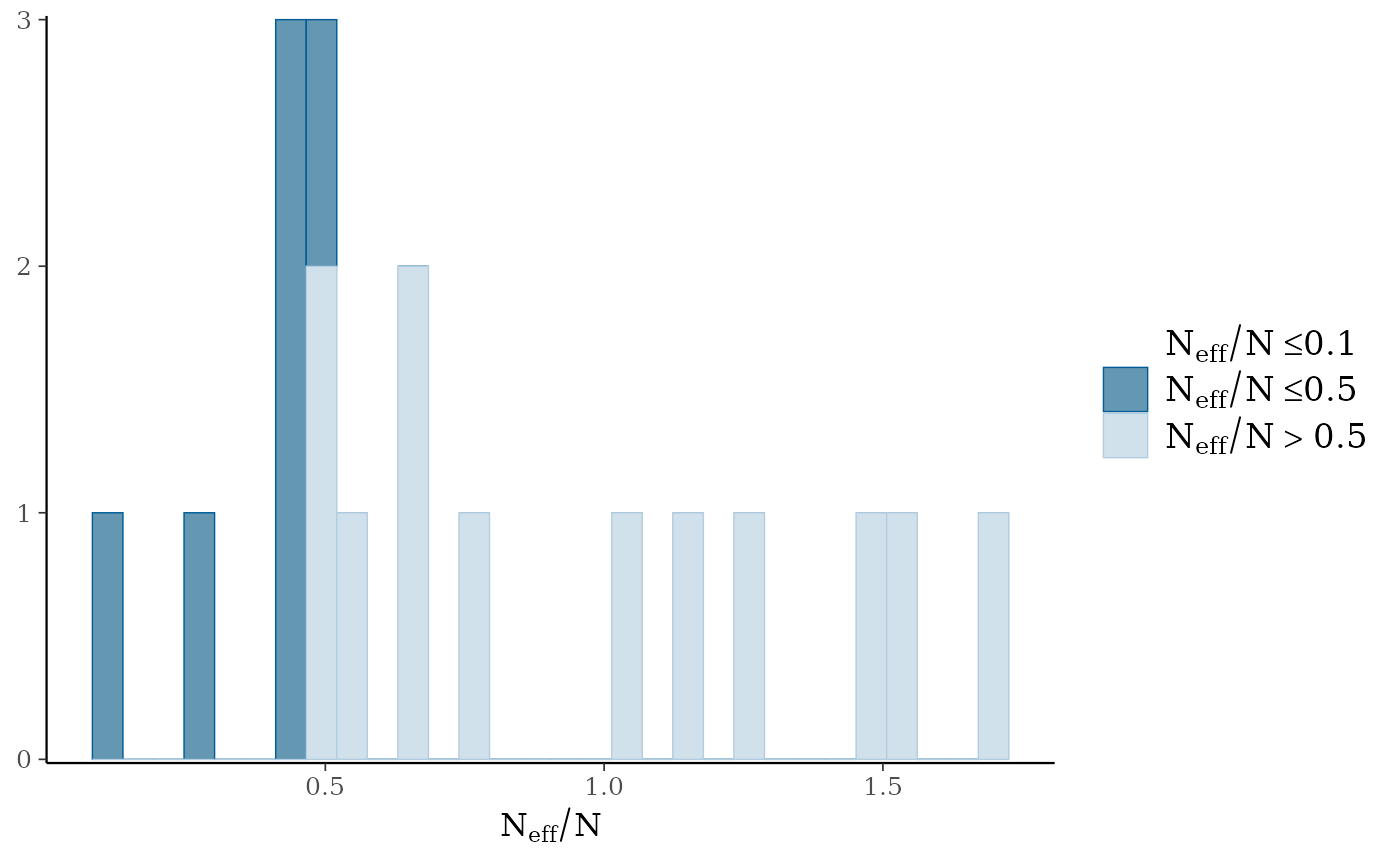
We can also examine the output for each parameter visually using a
traceplot combined with a density plot, which is given by the default
plot() command:
plot(mglmm_fit, ask = FALSE)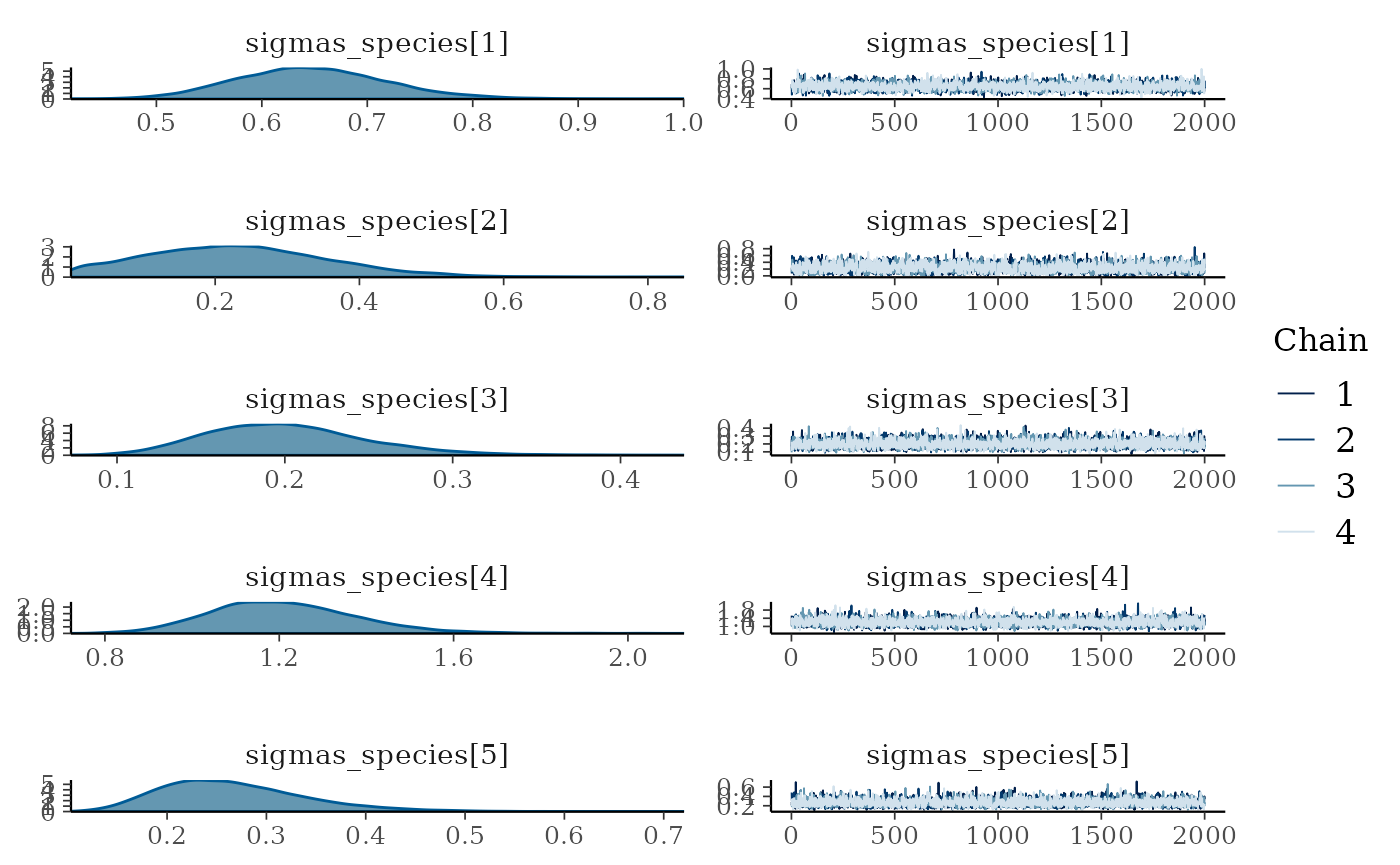
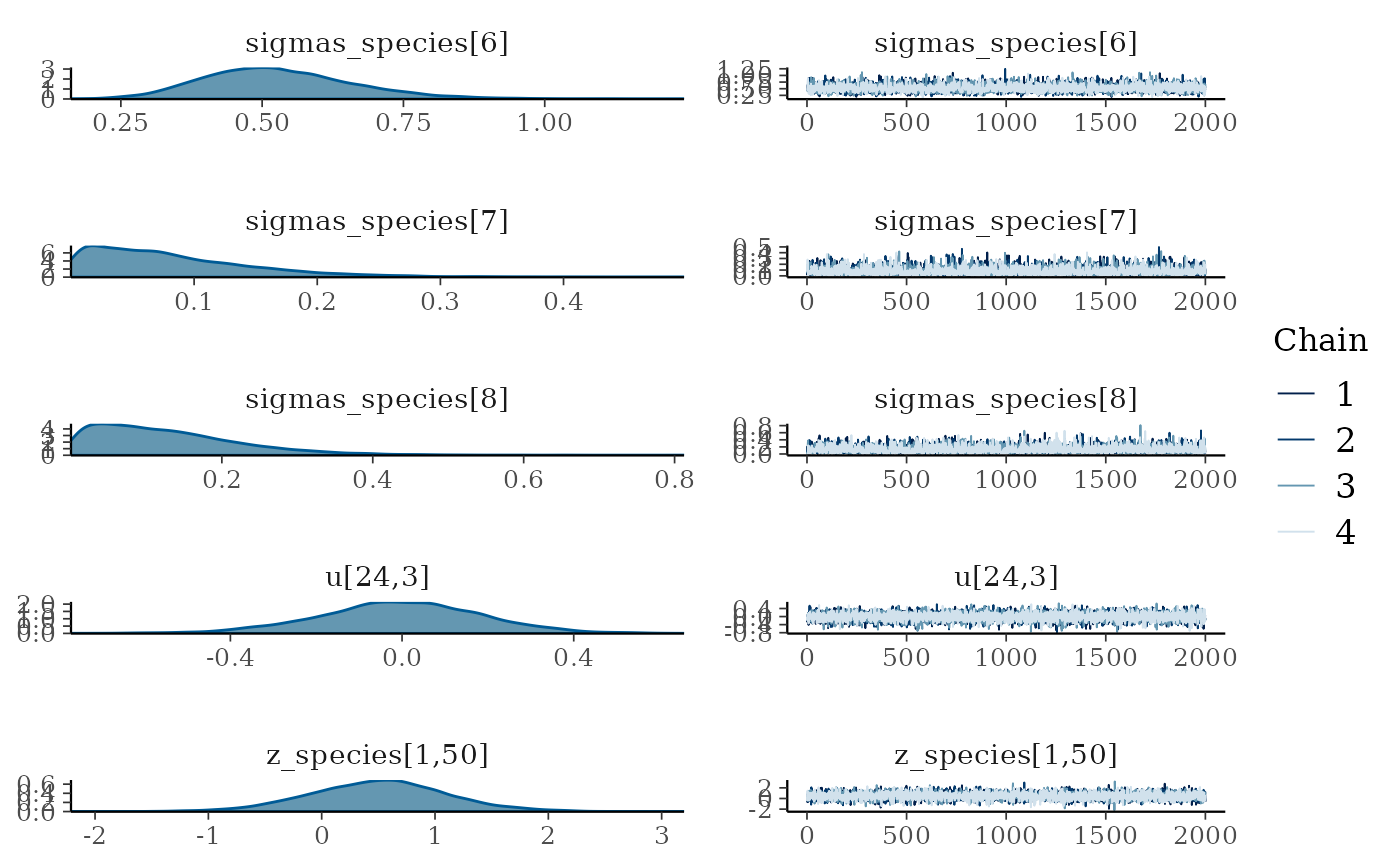
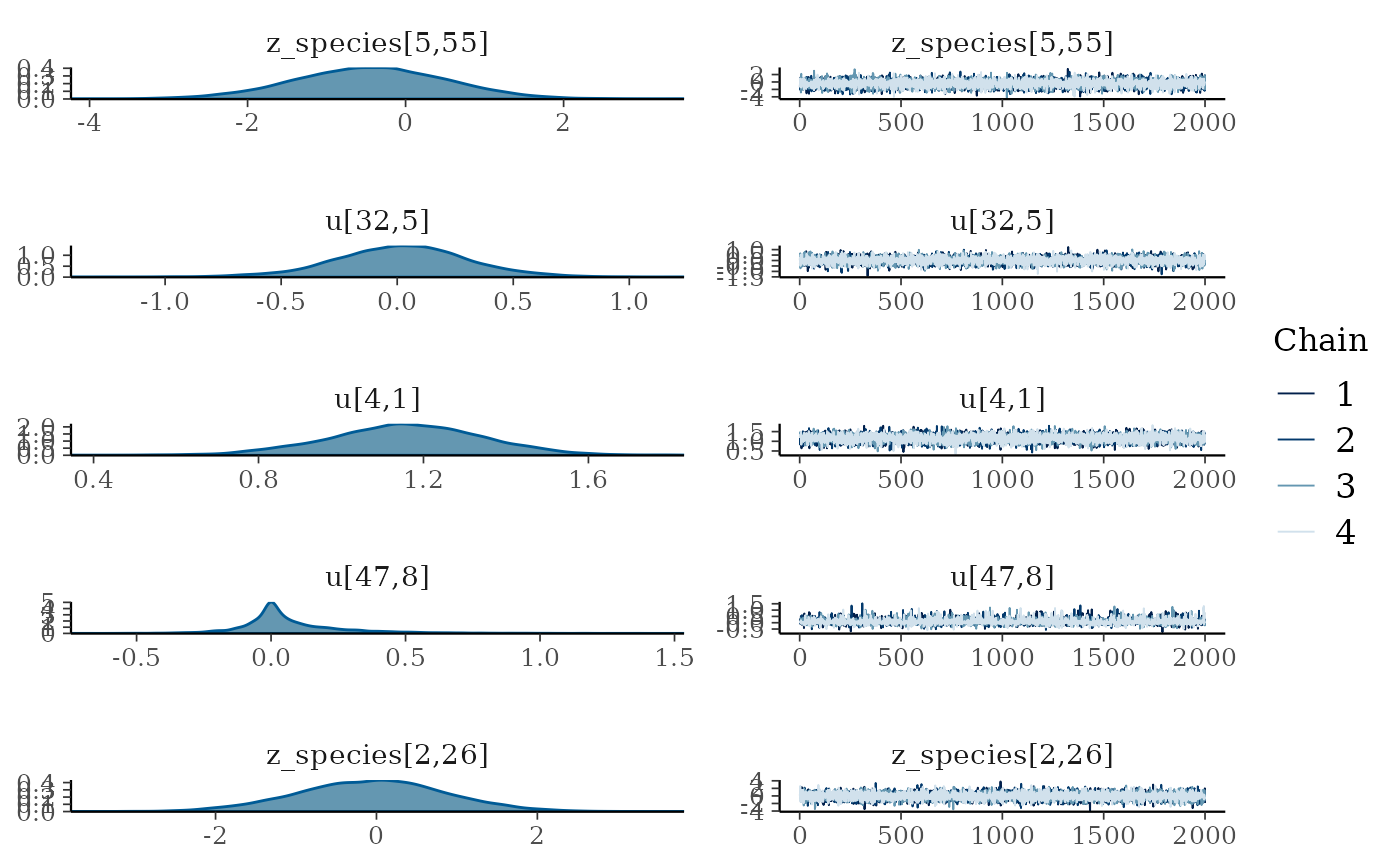
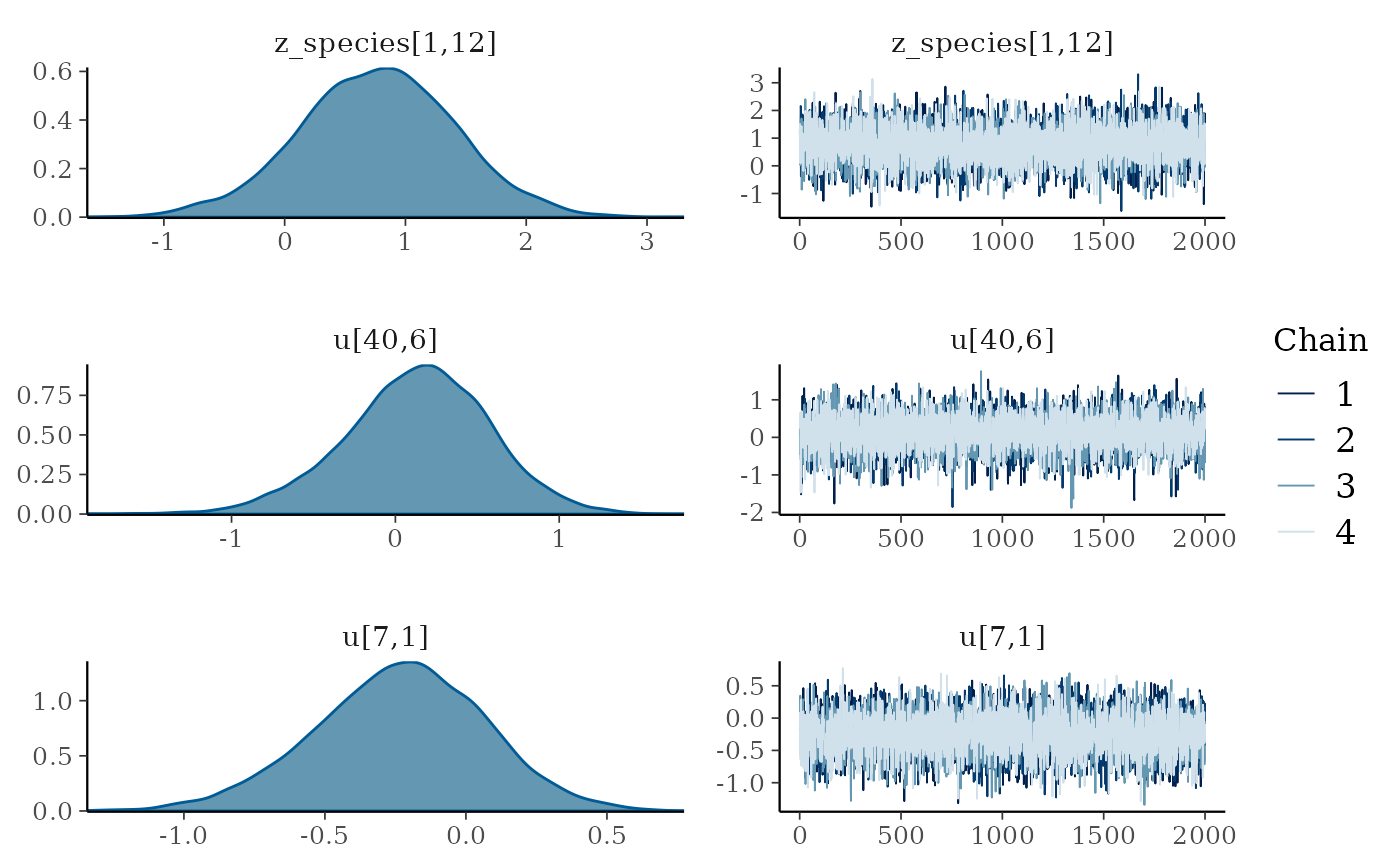
By default the plot() command plots all of the
parameters with sigma or kappa in their name plus a random selection of
20 other parameters, but this can be overridden by either specifying the
parameters by name (with or without regular expression matching) or
changing the number of parameters to be randomly sampled. Use the
get_parnames() function to get the names of parameters
within a model - and the jsdm_stancode() function can also
be used to see the underlying structure of the model.
All the mcmc plot types within bayesplot are supported by the
mcmc_plot() function, and to see a full list either use
bayesplot::available_mcmc() or run mcmc_plot()
with an incorrect type and the options will be printed.
We can also view the environmental effect parameters for each species
using the envplot() function.
envplot(mglmm_fit)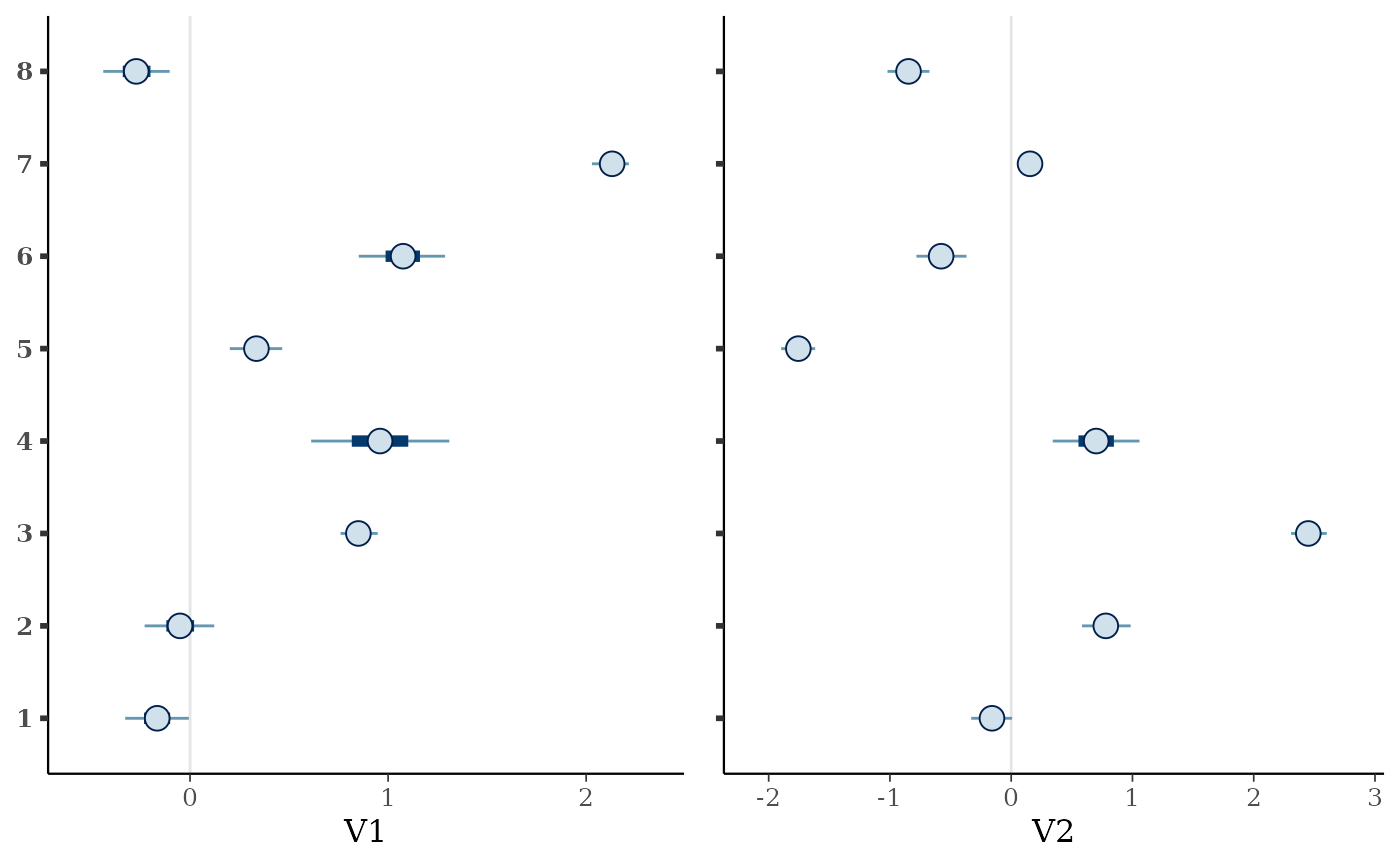
Posterior predictions can be extracted from the models using either
posterior_linpred() or posterior_predict(),
where the linpred function extracts the linear predictor for the
community composition within each draw and the predict function combines
this linear predictor extraction with a random generation based on the
predicted probability for the family. Both functions by default return a
list of length equal to the number of draws extracted, where each
element of the list is a sites by species matrix.
mglmm_pp <- posterior_predict(mglmm_fit)
length(mglmm_pp)
#> [1] 8000
dim(mglmm_pp[[1]])
#> [1] 75 8As well as the MCMC plotting functions within bayesplot the ppc_
family of functions is also supported through the
pp_check() function. This family of functions provides a
graphical way to check your posterior against the data used within the
model to evaluate model fit - called a posterior retrodictive check (or
posterior predictive historically and when the prior only has been
sampled from). To use these you need to have set
save_data = TRUE within the stan_jsdm() call.
Unlike in other packages by default pp_check() for
jsdmStanFit objects extracts the posterior predictions then
calculates summary statistics over the rows and plots those summary
statistics against the same for the original data. The default behaviour
is to calculate the sum of all the species per site - i.e. total
abundance.
pp_check(mglmm_fit)
#> Using 10 posterior draws for ppc plot type 'ppc_dens_overlay' by default.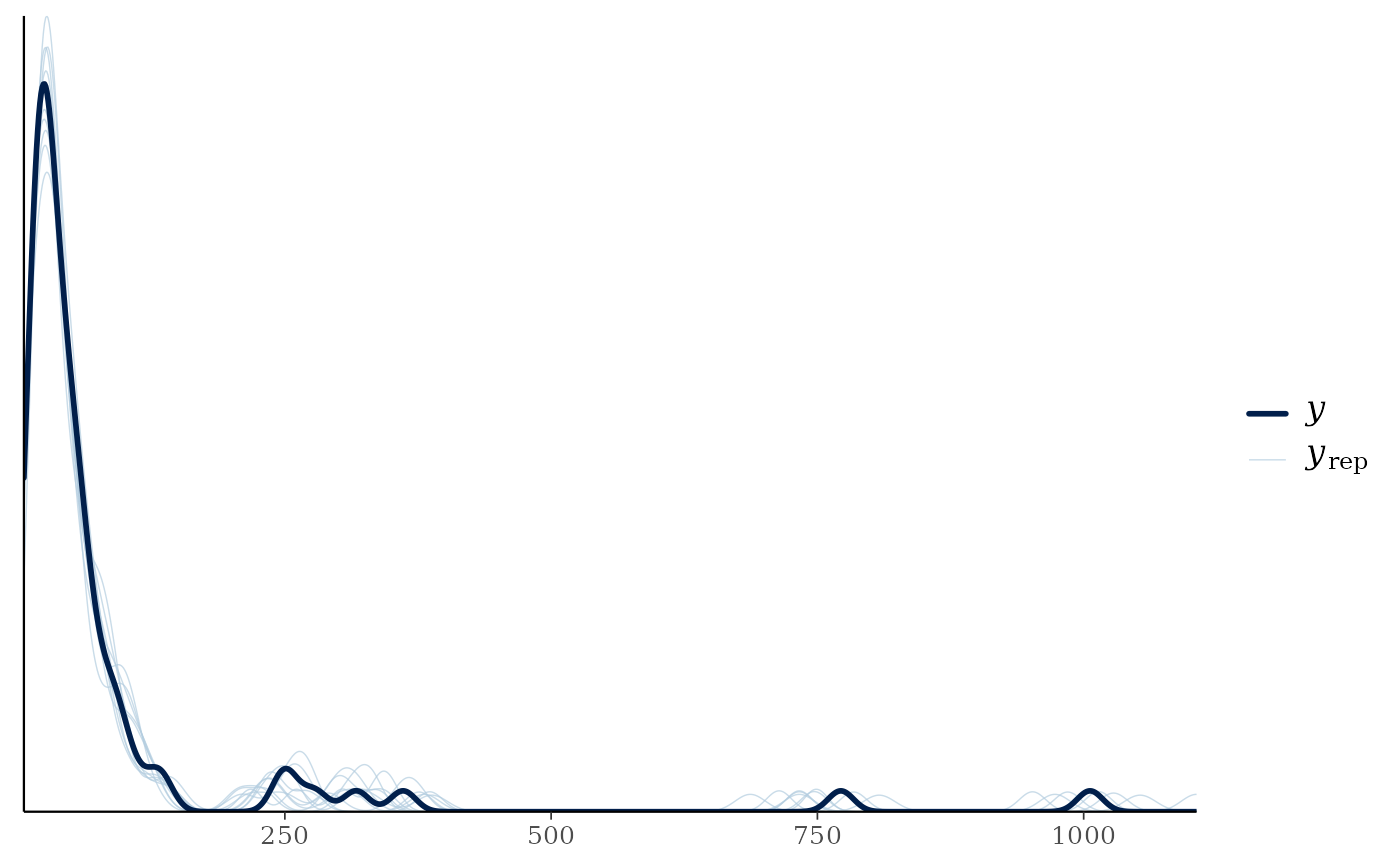
The summary statistic can be changed, as can whether it is calculated for every species or every site:
pp_check(mglmm_fit, summary_stat = "mean", calc_over = "species",
plotfun = "ecdf_overlay")
#> Using 10 posterior draws for ppc plot type 'ppc_ecdf_overlay' by default.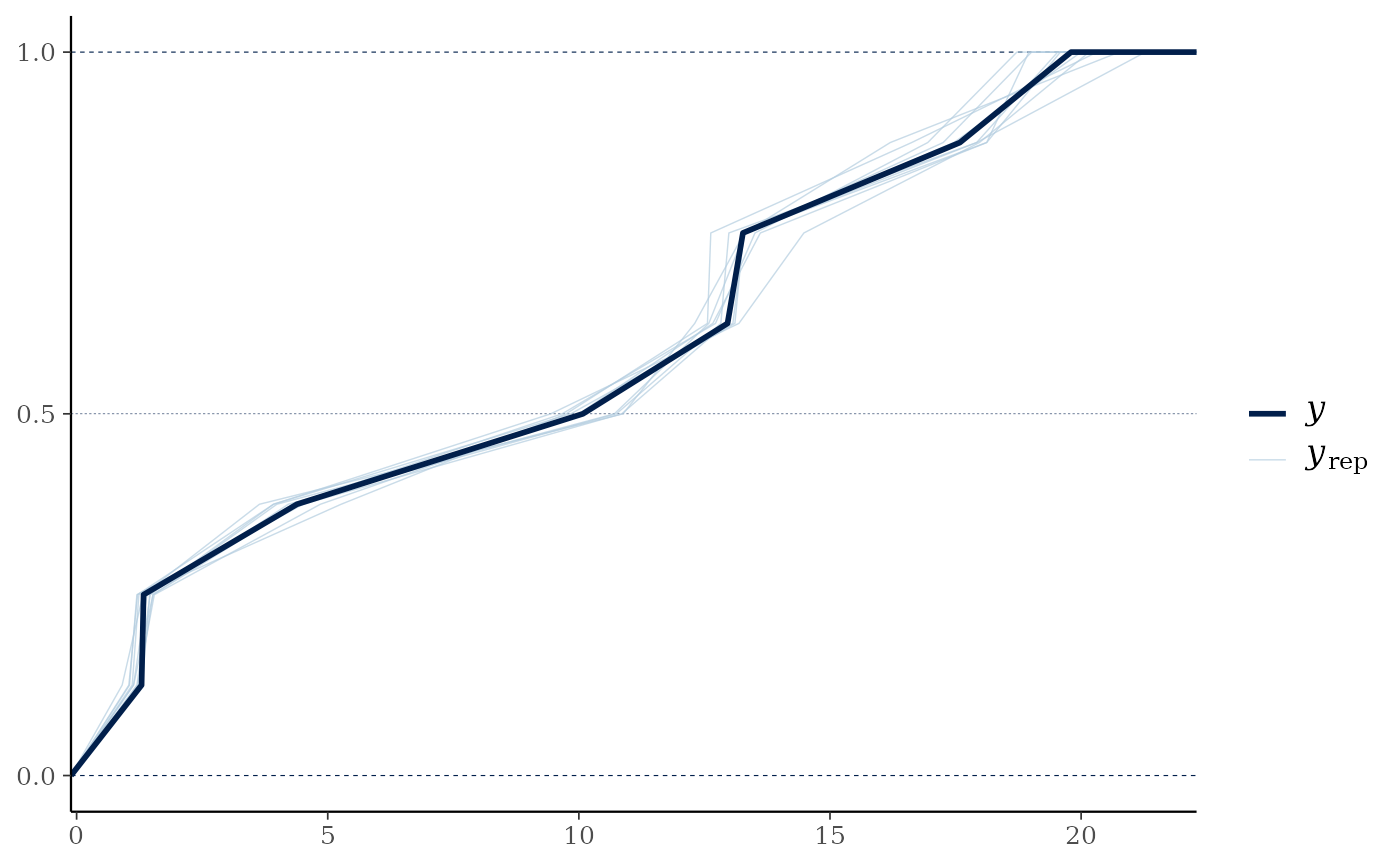
We can examine the species-specific posterior predictive check
through using multi_pp_check(), or examine how well the
relationships between specific species are recovered using
pp_check() with plotfun = "pairs".
As we have run the above model on simulated data and the original
data list contains the parameters used to simulate the data we can use
the mcmc_recover_ functions from bayesplot to
see how the model did:
mcmc_plot(mglmm_fit, plotfun = "recover_hist",
pars = paste0("sigmas_species[",1:8,"]"),
true = mglmm_test_data$pars$sigmas_species)
#> `stat_bin()` using `bins = 30`. Pick better value `binwidth`.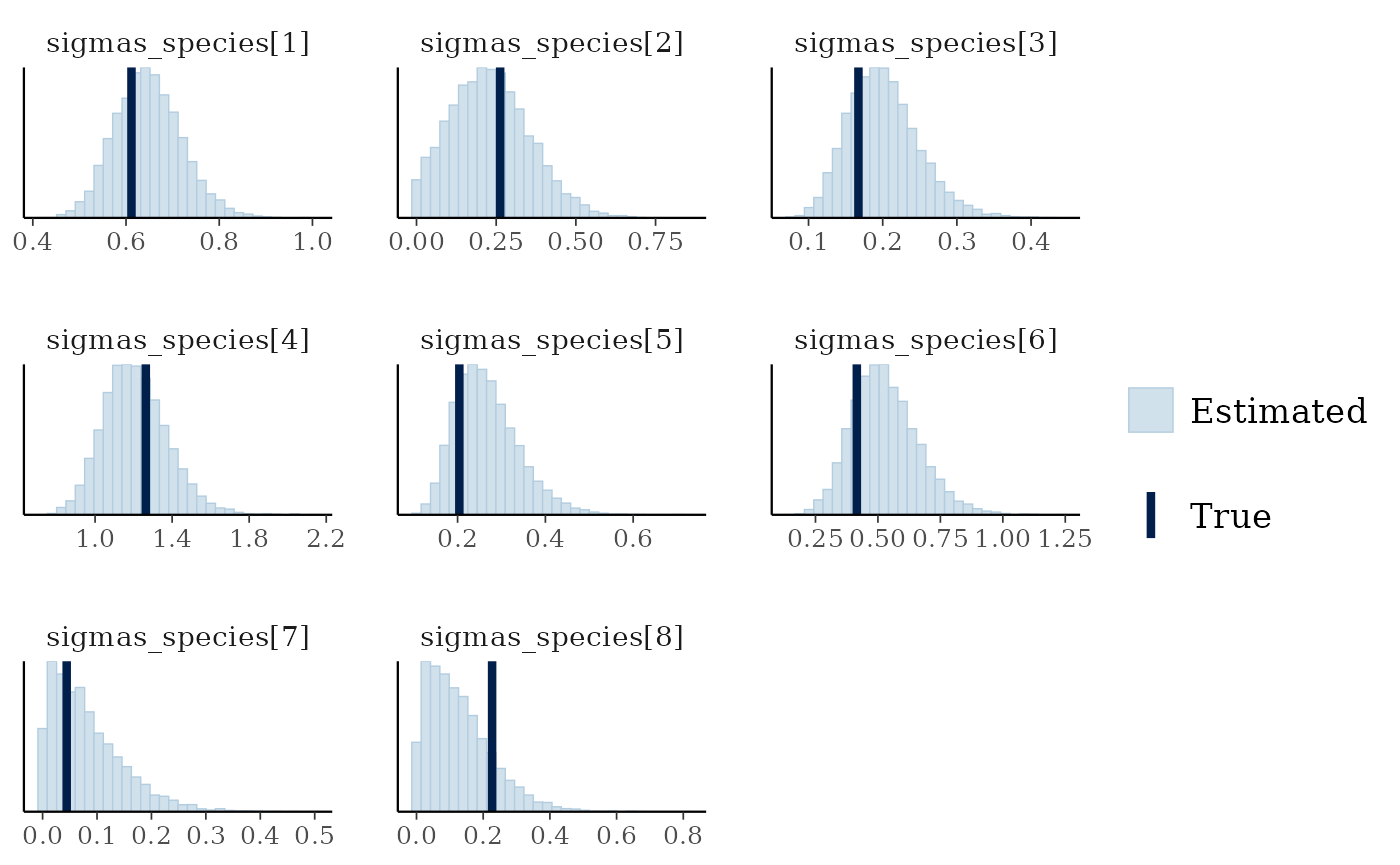
mcmc_plot(mglmm_fit, plotfun = "recover_intervals",
pars = paste0("cor_species[",rep(1:nspecies, nspecies:1),",",
unlist(sapply(1:8, ":",8)),"]"),
true = c(mglmm_test_data$pars$cor_species[lower.tri(mglmm_test_data$pars$cor_species, diag = TRUE)])) +
theme(axis.text.x = element_text(angle = 90))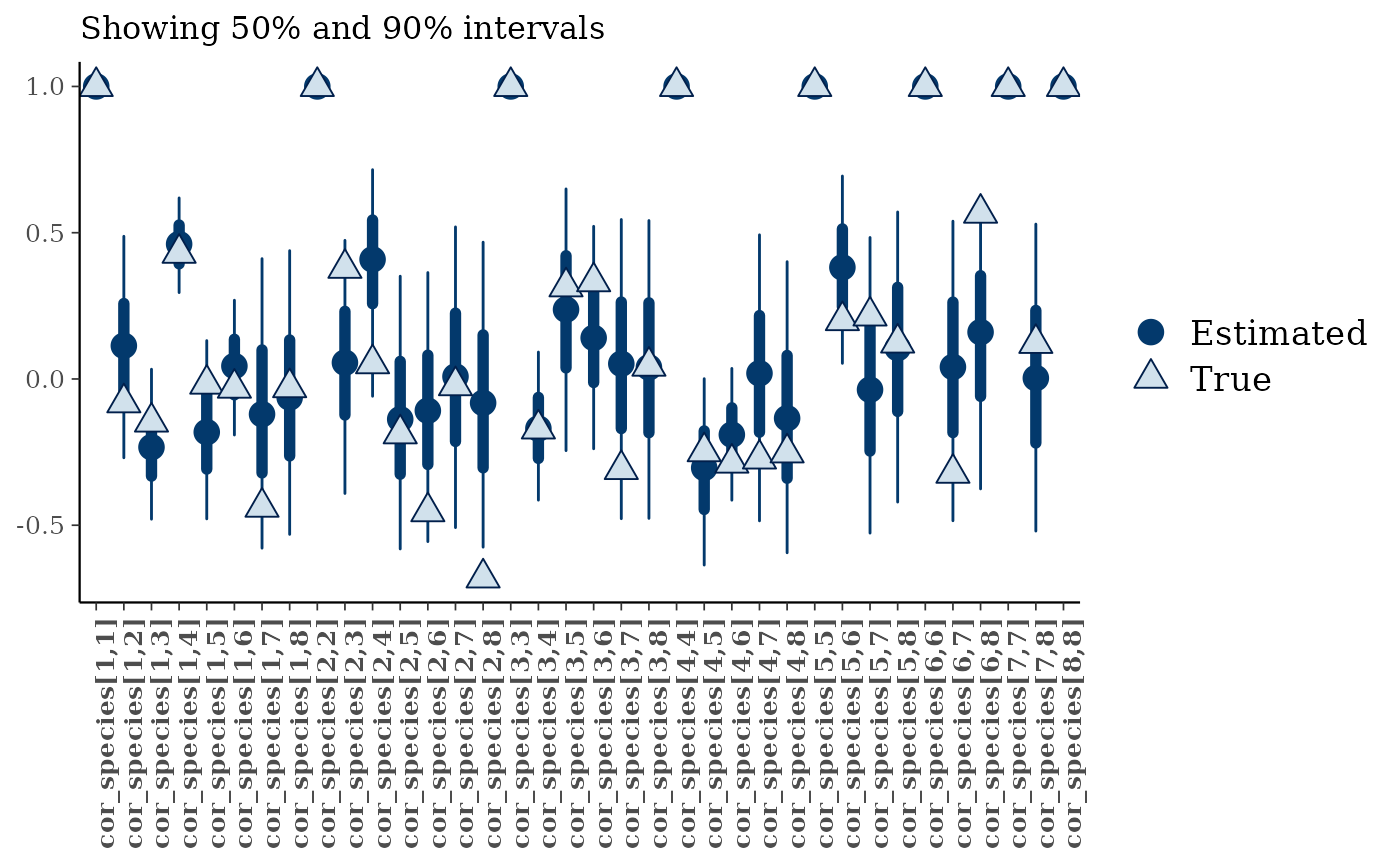
Fitting a GLLVM
The model fitting workflow for latent variable models is very similar to that above, with the addition of specifying the number of latent variables (D) in the data simulation and model fit. Here we change the family to a Bernoulli family (i.e. the special case of the binomial where the number of trials is 1 for all observations), make the covariate effects on each species draw from a correlation matrix such that information can be shared across species, and change the prior to be a Student’s T prior on the predictor-specific sigma parameter.
set.seed(3562251)
gllvm_data <- gllvm_sim_data(N = 50, S = 12, D = 2, K = 1,
family = "bernoulli",
beta_param = "cor",
prior = jsdm_prior(sigmas_preds = "student_t(3,0,1)"))
gllvm_fit <- stan_jsdm(Y = gllvm_data$Y, X = gllvm_data$X,
D = gllvm_data$D,
family = "bernoulli",
method = "gllvm",
beta_param = "cor",
prior = jsdm_prior(sigmas_preds = "student_t(3,0,1)"),
refresh = 0)
gllvm_fit
#> Family: bernoulli
#> Model type: gllvm with 2 latent variables
#> Number of species: 12
#> Number of sites: 50
#> Number of predictors: 0
#>
#> Model run on 4 chains with 4000 iterations per chain (2000 warmup).
#>
#> No parameters with Rhat > 1.01 or Neff/N < 0.05Again, the diagnostic statistics seem reasonable:
mcmc_plot(gllvm_fit, plotfun = "rhat_hist")
#> Warning: Dropped 1 NAs from 'new_rhat(rhat)'.
#> `stat_bin()` using `bins = 30`. Pick better value `binwidth`.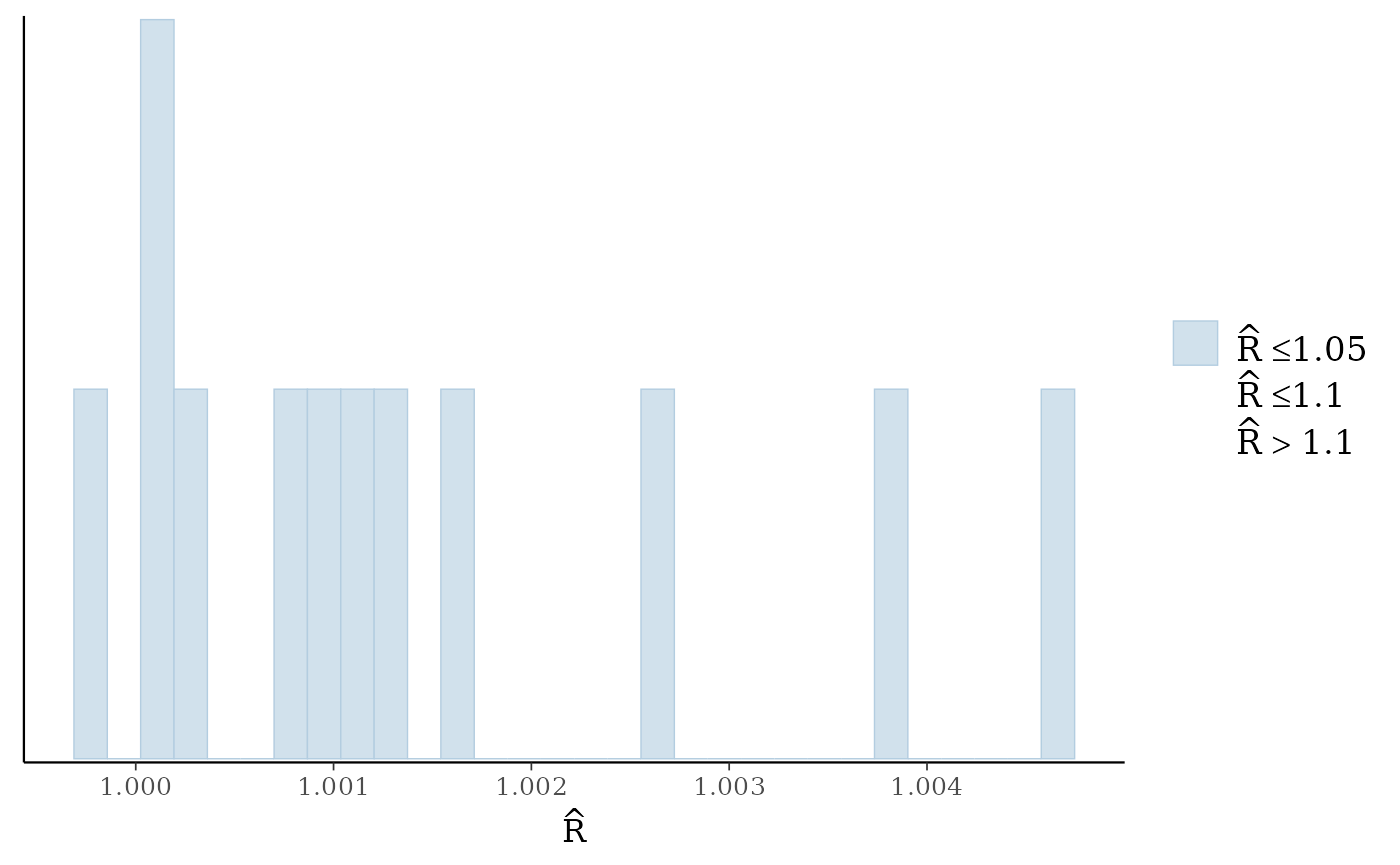
mcmc_plot(gllvm_fit, plotfun = "neff_hist")
#> `stat_bin()` using `bins = 30`. Pick better value `binwidth`.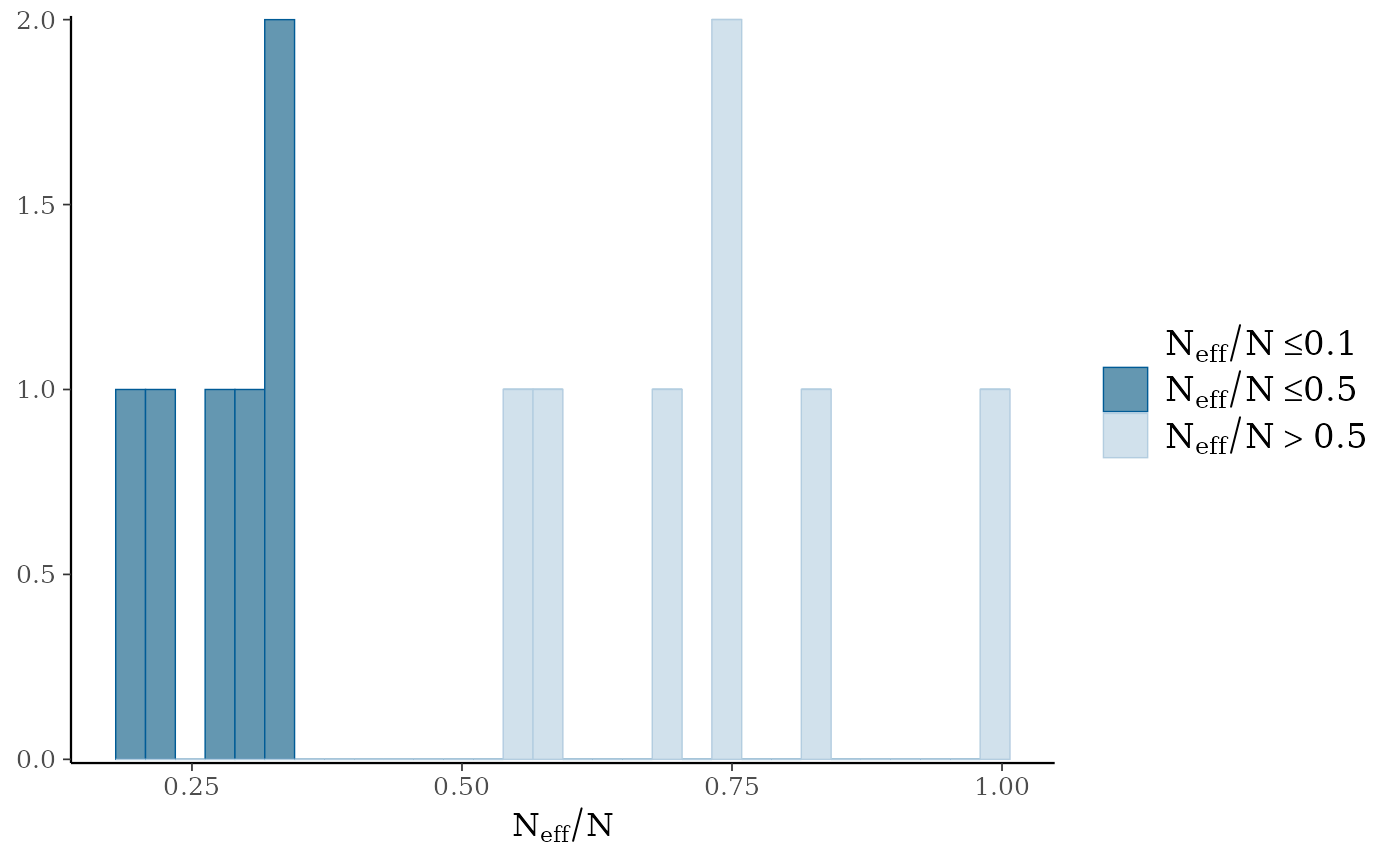
For brevity’s sake we will not go into the detail of the different
functions again here, however there is one plotting function
specifically for GLLVM models - ordiplot(). This plots the
species or sites scores against the latent variables from a random
selection of draws:
ordiplot(gllvm_fit, errorbar_range = 0.5)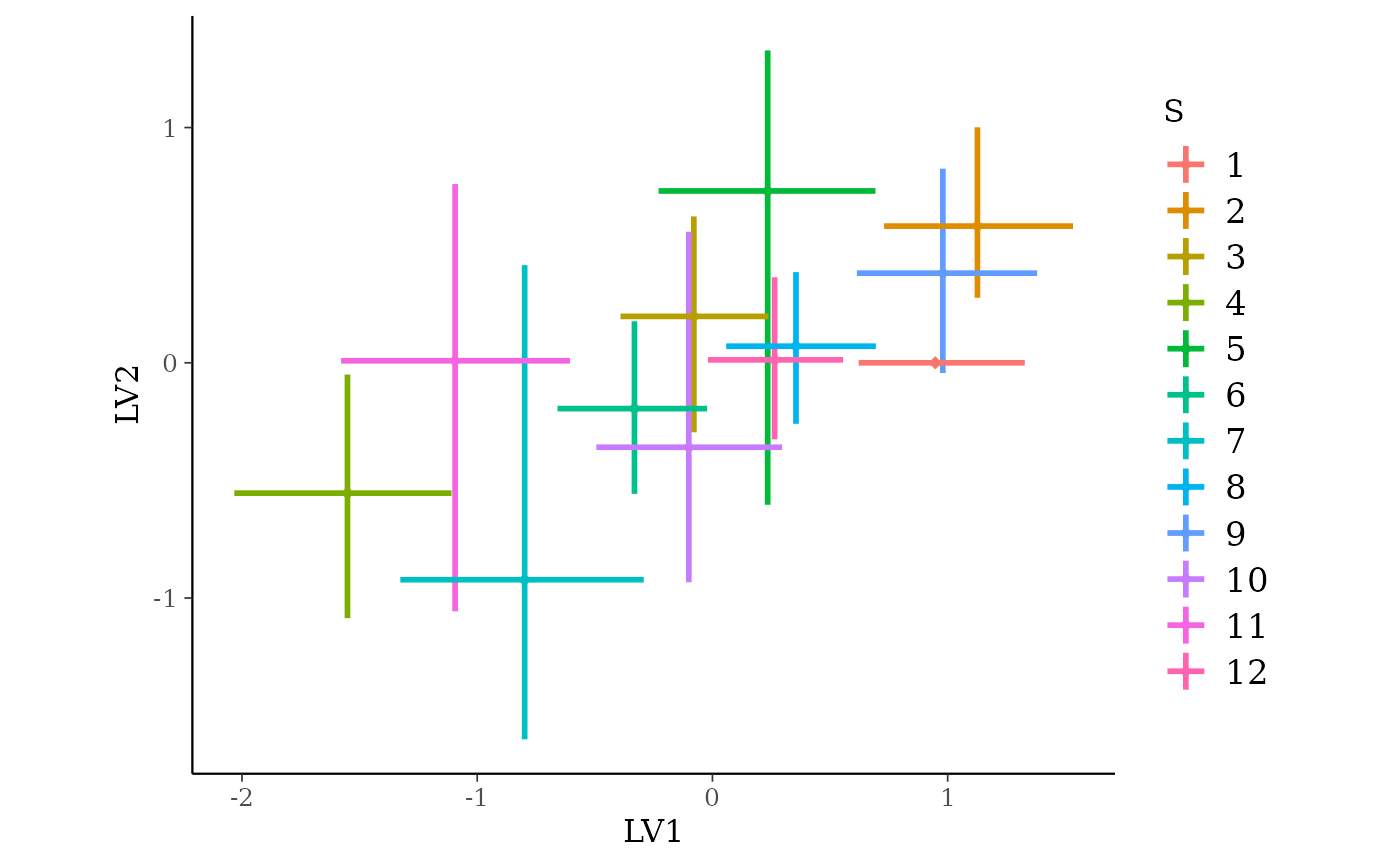
ordiplot(gllvm_fit, type = "sites", geom = "text", errorbar_range = 0) +
theme(legend.position = "none")
You can change the latent variables selected by specifying the
choices argument, and alter the number of draws or whether
you want to plot species or sites with the other arguments.
References
Warton et al (2015) So many variables: joint modeling in community ecology. Trends in Ecology & Evolution, 30:766-779. DOI: 10.1016/j.tree.2015.09.007.
Wilkinson et al (2021) Defining and evaluating predictions of joint species distribution models. Methods in Ecology and Evolution, 12:394-404. DOI: 10.1111/2041-210X.13518.
Vehtari, A., Gelman, A., and Gabry, J. (2017). Practical Bayesian model evaluation using leave-one-out cross-validation and WAIC. Statistics and Computing. 27(5), 1413–1432. DOI: 10.1007/s11222-016-9696-4.